Python
GFF3 Parser
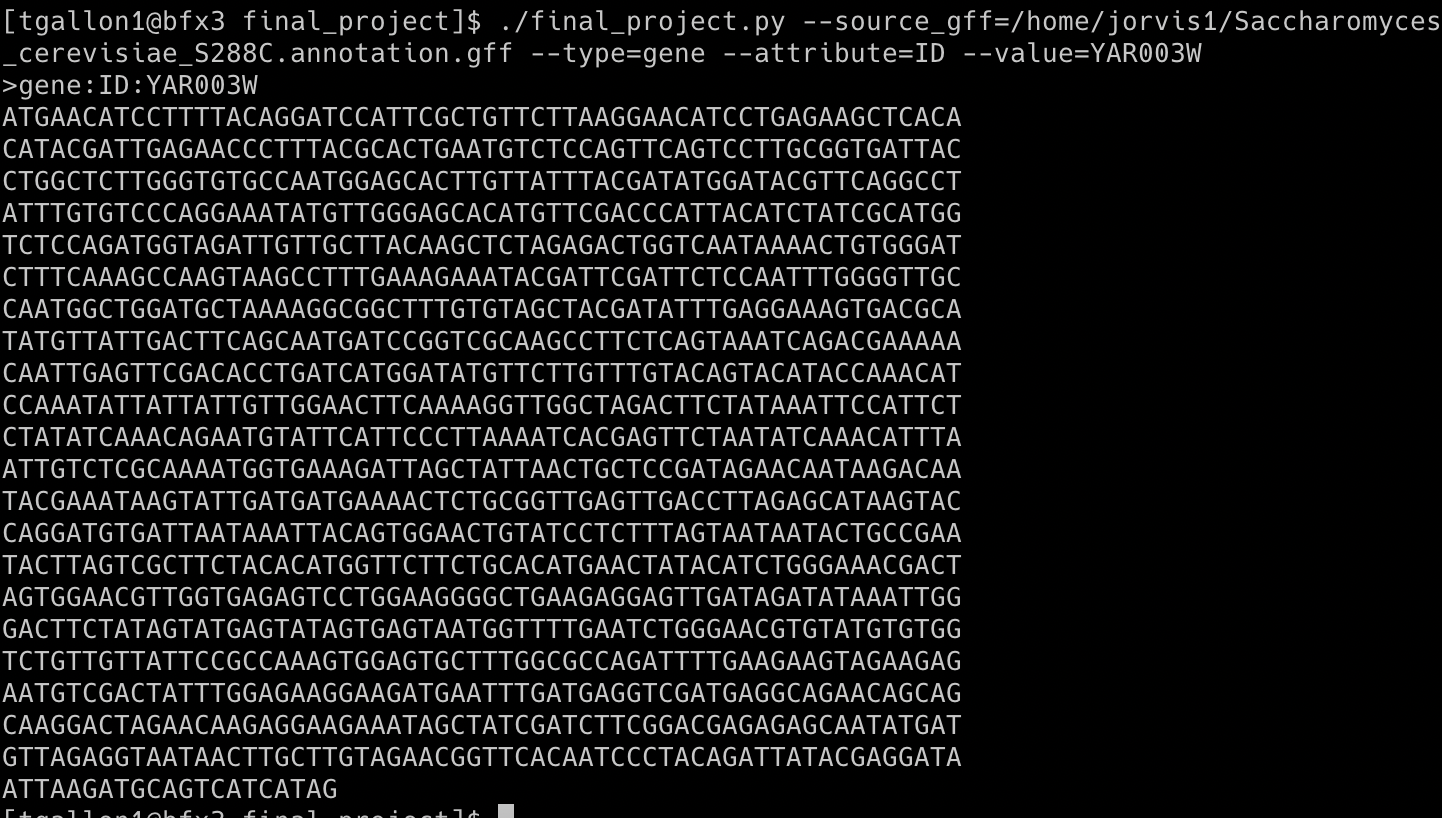
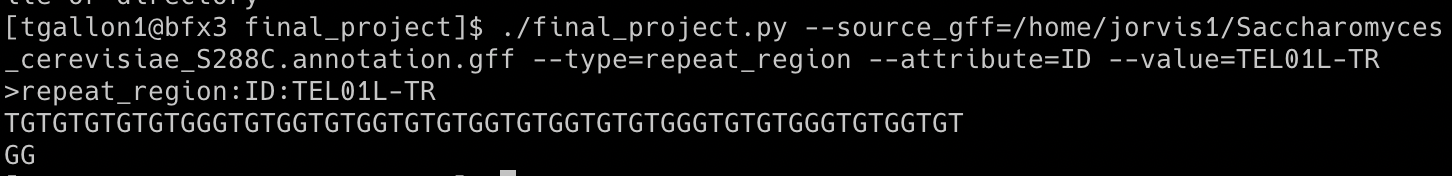
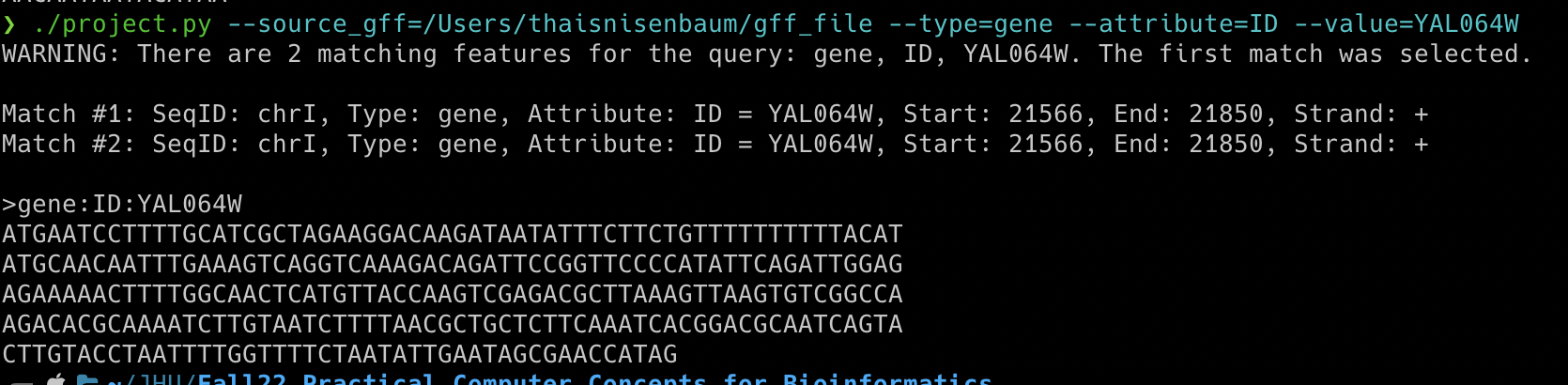
GFF3 files are nine-column, tab-delimited, plain text files used for sequence annotations. I wrote a GFF3 feature exporter that takes in four arguments corresponding to values in the GFF3 columns. Below is an example of how to run the script on the command line, which reads the path to a GFF3 file and finds any feature type (column 3: type) with a specified ID (column 9: attributes). Then, the script identifies the sequence coordinates associated with the specified input (columns 4: start, 5: end, and 7: strand) and prints the FASTA sequence as 60 characters per line, which follows the standard. Below are example outputs using the genome annotation file for Saccharomyces cerevisiae S288C (Saccharomyces_cerevisiae_S288C.annotation.gff)
Example Output 1: FASTA sequence of the gene with an ID of YAR003W
Example Output 2: FASTA sequence of a repeat region with an ID of TEL01L-TR
Example Output 3: Output if the input value is not in the file

Example Output 4: Output if the file contains duplicated values
More information about the GFF3 format can be foundhere.
BLASTN File Parser
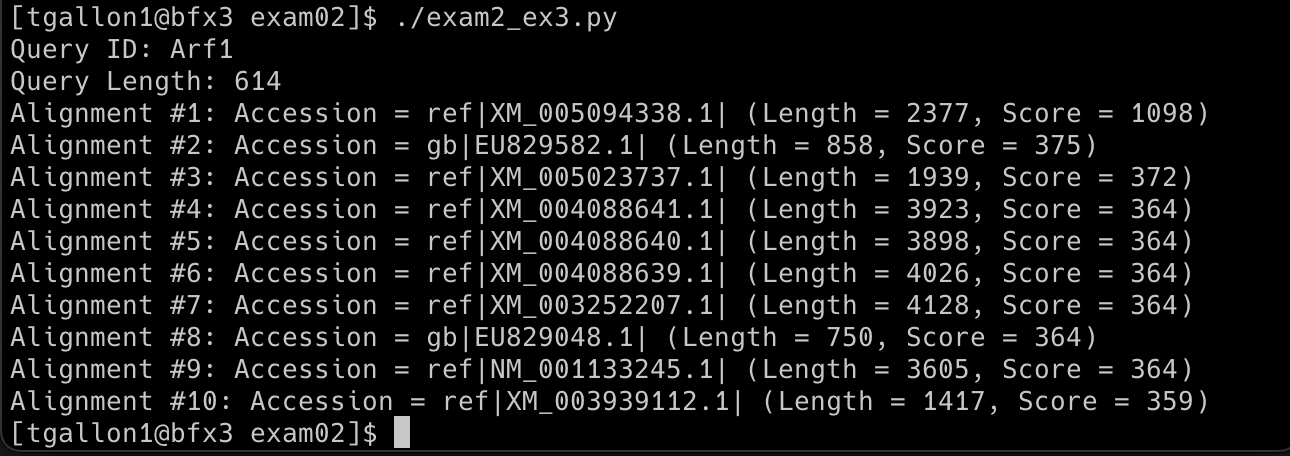
I wrote a program that opens a BLASTN (nucleotide to nucleotide search) output file, parses out specific information, and produces a formatted output. The output displays information about the query sequence ID and query length, then it parses information about the significant alignments for this query and displays the first ten hits containing accession information, length, and score.
Output:
Conversion of a GFF3 File to a BED File
I wrote a Python script that transforms the gene features of a GFF3 formatted file to BED format.
The example below uses the GFF3 file of the Saccharomyces cerevisiae S288C genome, which contains a total of 384 genes.
Output printed to the console, as shown below, and written to yeast.bed file including chrom, chromStart, chromEnd, name, score, and strand.
Specifications for BED format can be foundhere.